As summer melts into fall, informed consent is on the line, with FDA approval of the Pfizer vaccine expected by Labor Day. Full approval will usher in changes that will impact our rights. There is no question about this. The question is, though, what can we do about it?
We have seen sporadic COVID vaccine mandates by employers, schools, and governments, but many are holding off. The legal footing for mandates is built on sand but still more solid when vaccines are approved rather than authorized. Vaccine approval will trigger mandates across the country in the U.S. military, entire cities, the federal government, college campuses, and more. Education and employment are already being jeopardized by the jab as condition of access. Pfizer’s impending FDA approval has public and private bodies rushing to use mandates to fortify the castle.
Things are changing rapidly. It’s time to draw our line in the sand.
What does full approval mean?
Full approval eliminates EUA red tape for COVID vaccine use. Under full approval, a doctor can use discretion in off-label uses like boosters or administrations to younger children. The FDA will grant no more vaccine EUAs because there is a “safe and effective” product on the market. Approval should trigger an end to the EUAs for Moderna and Janssen vaccines, since there would be an approved alternative.
Former FDA chief scientist Jesse Goodman asserts approval and experimental authorization is “not a huge difference, but it is a real difference.” i Other officials have concurred, “[It] is just a matter of degree.” ii
On one hand, they may be right. The FDA approval for the vaccines will not change anything about the science of it, nor does it appear the integrity of the data will be called into question, and Pfizer shots will continue to be injected under a different legal framework.
But on the other hand, the legal differences are like a tsunami that could wipe out human rights in America.
Our bodies and the law
There is no space more sacred than our bodies. This truth is enshrined in American law.
“No right is held more sacred, or is more carefully guarded by the common law, than the right of every individual to the possession and control of his own person, free from all restraint or interference of others, unless by clear and unquestionable authority of law.” This notion of bodily integrity has been embodied in the requirement that informed consent is generally required for medical treatment.” iii (emphasis added) -Supreme Court Justice William Rehnquist
American citizens hold dear the constitutionally protected right to make informed choices about medical interventions. These natural laws and human rights have been reaffirmed by our Supreme Court time and time again.
It is on these principles that America’s Frontline Doctors filed a lawsuit praying to halt not only EUAs, but also full approval, citing faulty CDC data about safety and efficacy. Without trustworthy data, Americans cannot truly give informed consent, nor properly exercise their right to refusal.
Most Americans do not know how a drug becomes “safe and effective,” yet that information is crucial to assessing its risks and benefits. Here’s a peek at the process.
[/et_pb_text]
The usual path to vaccine approval
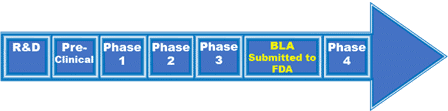
Typical FDA vaccine approval process. iv
The above graphic shows the typical FDA vaccine approval process, which generally takes 10-15 years. Lab research (up to 10 years of the process) precedes pre-clinical animal testing. Next, 20–100 healthy, unexposed humans are recruited for Phase 1 studies of immune response and dosing. Phase 2, Randomized Control Trials, includes hundreds of people of mixed health, age, and ethnicity, expanding to thousands for Phase 3. Safety is the primary focus early in the process, which gives way to a focus on efficacy and strategy for scalable manufacture. At this point, the vaccine candidate would have enough data to submit a Biologics License Application (BLA). Until the BLA is approved, the vaccine is experimental.
Operation Warp Speed, EUAs, and priority approval of BLA
The fastest vaccine development prior to COVID was mumps, in 4 years. COVID vaccines were authorized under EUAs approximately a year from the first known case of the virus, spurred on by the funding and focus of Operation Warp Speed.
The Secretary of Health and Human Services declared a public health emergency triggering the use of EUAs on January 31, 2020. On March 13, 2020, former President Trump declared a national emergency, unlocking funding for states and expanding executive power. Four days later, on March 17, Pfizer announced its intent to develop a COVID vaccine with partner BioNTech. The companies had begun research three months earlier, after publication of the virus genome.
Pfizer’s vaccine is on track to be fully FDA approved as early as Labor Day this year, and thus will be the focus for this article.
Betting on gene-based tech
“To save time the researchers took unorthodox steps.” v
Pfizer and BioNTech, a German company specializing in mRNA technology, partnered in 2018 to make flu shots with mRNA technology. While these haven’t come to market yet, Pfizer looked to mRNA as a strategy for COVID vaccine rapid development. vi “Pfizer was racing to develop a radical new vaccine based on a technology that had never been approved before,” as reported in the Wall Street Journal. Software was used to design the candidates, rather than lengthy test-tube cultivation. Novel technology and prior research on mRNA delivery shaved time off the research phase.
Pfizer’s head of vaccine research, Dr. Katherin Jansen, (co-developer of Merck’s Gardasil) led a team to select a candidate from 20 potential vaccines. By mid-April 2020, four options were selected for human testing. In the interest of speed, Pfizer started human trials simultaneously with animal trials, an unprecedented decision.
Combining phases, testing multiple vaccine candidates simultaneously, manufacturing before authorization
Phase 1 started April 23, 2020, on human volunteers in Germany, which immediately eliminated two candidates due to reactions. Ultimately, Pfizer settled on moving forward with the vaccine Americans are using today because it produced “fewer cases of fevers and chills,” and was thus “more tolerable.”
FDA approval requires a plan for scalable manufacture. “One of the ideas for speeding the process was to manufacture “at risk” – to start making product even before it had been proven safe and effective.” vii Because the company did not have time to test how long doses could be refrigerated, they chose to ship at sub-arctic temperatures. Pfizer declined federal money for its production network, because “they didn’t want to give agencies outside the FDA more leverage over the design of the trials.” This gamble paid off, as Moderna’s use of federal money slowed their process when officials required more racial and ethnic diversity of study subjects. Pfizer began manufacturing the vaccine in mid-August 2020, three months before an EUA was granted.
Pfizer also combined trials in Phases 2 and 3, which does occur occasionally in normal drug development. In the U.S., the first volunteers were injected in New York on July 27, 2020.
The company had unexpected trouble enrolling patients. “[F]ewer subjects than expected had become sick,” perhaps due to health precautions or maybe, questioned lead scientist Dr. Jansen, “the FDA-authorized tests Pfizer was using to confirm cases weren’t accurate.”
Somehow the issue resolved itself and enough cases were confirmed. (You can dive deeper into the trials in a paper published by Dr. Henry Ealy with GreenMedInfo, on page 153 – entire 444 page document).
On November 20, 2020, Pfizer submitted an EUA for approval of their vaccine candidate BNT162b2. viii Phase 3 trials were ongoing.
On May 7, 2021, Pfizer had enough data to submit a BLA for full FDA approval. Pfizer was granted priority review, giving the FDA only six months to issue a decision.
Where does that leave us?
Are COVID vaccines experimental regardless of legal status? Some say that isn’t true, but history and a logical reading of laws and court cases show they are.
A drug is considered experimental until it is granted full FDA approval for the specified intended use after three phases of clinical trials with specific timeframes for collection of safety data. Doctors may use drugs in off-label ways, but the drug cannot be marketed as such as it has not been vetted by clinical trials and FDA analysis. If the manufacturer wants to market the drug beyond what has been approved, that use must also go through the trials and approval process. ix
Emergency Use Authorizations (EUAs) were created to bypass FDA approval laws
COVID vaccines are the second types of vaccinations to be authorized for emergency use, but the first to be developed in tandem with authorization. In fact, the EUA law was created for off-label use of the Anthrax vaccine by the military. In 2004, in response to losing a court case where service members objected to off-label mandates, then-President George W. Bush signed into law Project Bioshield, bypassing the court ruling upholding FDA approval law, creating EUAs in the process. x xi The DOD was then able to administer the Anthrax vaccine off-label (approval was for skin absorption, not inhalation). xii
The FDA had 64 years of data — 30 years of research and 34 years post-market — to scrutinize the Anthrax vaccine before granting off-label temporary approval.
The right to refuse is not optional
Emergency Use Authorizations, experimental treatments, and informed consent require an option to refuse. EUA law dictates a recipient understand “the option to accept or refuse administration of the product,” along with the “consequences, if any, of refusing.” xiii Experimental drugs are governed by code requiring extensive and heightened informed consent. And SCOTUS has noted, “The logical corollary of the doctrine of informed consent is that the patient generally possesses the right not to consent, that is, to refuse treatment.” xiv
Arguments that COVID vaccines are not experimental is an attack on informed consent and right to refuse treatment. They are legally and logically unsound. The word “experimental” has become politicized to draw our attention away from the tenuous data upon which authorization rests. In asserting the vaccine is not experimental, the government is attempting to bypass the laws that require heightened informed consent by obscuring the risks. xv
When individuals are recruited for trials, the law requires certain information be disclosed and comprehended for consent to be considered fully informed. xvi The law also requires an IRB be “particularly cognizant of the special problems of research that involves a category of subjects who are vulnerable to coercion or undue influence, such as children, prisoners, individuals with impaired decision-making capacity, or economically or educationally disadvantaged persons.” xvii
The FDA claims the option requirement is satisfied by the vaccine fact sheets which state, “It is your choice to receive or not receive the … vaccine. Should you decide not to receive it, it will not change your standard medical care.” xviii
The Department of Justice asserted the option to refuse was itself an option for the government to offer and exercising the option could be met with serious consequences. (See SHF’s recent post, Is the government weaponizing data?) A Texas court ruled that an employer mandate could stand because the employee had the option to refuse. The Plaintiff asserted she was being forced to take the jab or be fired. The court responded, “This is not coercion … [She] can freely choose to accept or refuse a COVID-19 vaccine; however, if she refuses, she will simply need to work somewhere else.” xix
This my-way-or-the-highway governance feels more like extortion and less like policy. Both the DOJ and the Texas court conspicuously neglected constitutional analysis. If removal of constitutional rights is held as the consequence of noncompliance, consent dissolves into coercion.
The vaccine experiment is happening now, and we are all part of the trial. It’s not just about the novel genetic drug, but about our civil rights. How far can the government push our rights away before our grip on them is gone? The Constitution is being bypassed and our resolve in standing up for informed consent and choice in medical treatment is being tested right now.
Steps you can take
Step One: Tell your governor and local legislators that COVID-19 vaccines must be voluntary!
Step Two: Stand with the Frontline Doctors by asking your legislators to call for an investigation into CDC data. Without trustworthy and replicable data, we cannot make informed decisions about our health.
Hidden Toggle
References & Sources
ii https://www.aarp.org/health/conditions-treatments/info-2021/full-fda-approval-covid-vaccine.html
iii Cruzan, quoting Union Pacific R. Co. v. Botsford, 141 U. S. 250, 141 U. S. 251 (1891).
iv Image from https://www.fda.gov/vaccines-blood-biologics/development-approval-process-cber/vaccine-development-101
vi Combination Covid and Flu mRNA vaccines are already in the R&D pipeline. See https://www.clinicaltrialsarena.com/analysis/mrna-flu-vaccines/
viii https://www.fda.gov/media/144416/download
ix Gardasil 9 is a familiar example-the vaccine was licensed for females of a certain age range, licensed again for males, again to expand the age of use, and again for additional cancers.
x https://www.courtlistener.com/opinion/2459105/doe-v-rumsfeld/
xi https://www.healthaffairs.org/do/10.1377/hblog20210212.410237/full/
xii DoD was still required by law to obtain informed consent, and did not mandate the vaccine until full FDA approval terminated the EUA. We are seeing a similar pattern today with Covid vaccines, as the military has announced a mandate post-licensure.
xiii The HHS Secretary must establish appropriate conditions designed to ensure that individuals to whom the product is administered are informed
(I) that the Secretary has authorized the emergency use of the product;
(II) of the significant known and potential benefits and risks of such use, and of the extent to which such benefits and risks are unknown; and
(III) of the option to accept or refuse administration of the product, of the consequences, if any, of refusing administration of the product, and of the alternatives to the product that are available and of their benefits and risks. (emphasis added)
xiv Cruzan v. Director, MO Dept of Health, 497 US 261 (1990)
In experimental treatment, citizens arguably have heightened consent rights. Debate over the Right to Try Act (RTT), which put patients in direct contact with experimental drug manufacturers outside of clinical trials, was product, in part, of the extreme level of precaution asserted by our government over experimental drugs. Critics lambasted RTT for cutting out Institutional Review Boards (IRBs), claiming the committees were the safeguard of informed consent. The assumption was IRBs would protect against unscrupulous doctors and companies who might try to sell an ineffective or harmful drug to a vulnerable patient.
xvi 21 CFR 50.20, 50.25 https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=50.20; https://ecfr.io/Title-21/Section-50.25
§ 50.25 Elements of informed consent.
(a) Basic elements of informed consent. In seeking informed consent, the following information shall be provided to each subject:
(1) A statement that the study involves research, an explanation of the purposes of the research and the expected duration of the subject’s participation, a description of the procedures to be followed, and identification of any procedures which are experimental.
(2) A description of any reasonably foreseeable risks or discomforts to the subject.
(3) A description of any benefits to the subject or to others which may reasonably be expected from the research.
(4) A disclosure of appropriate alternative procedures or courses of treatment, if any, that might be advantageous to the subject.
(5) A statement describing the extent, if any, to which confidentiality of records identifying the subject will be maintained and that notes the possibility that the Food and Drug Administration may inspect the records.
(6) For research involving more than minimal risk, an explanation as to whether any compensation and an explanation as to whether any medical treatments are available if injury occurs and, if so, what they consist of, or where further information may be obtained.
(7) An explanation of whom to contact for answers to pertinent questions about the research and research subjects’ rights, and whom to contact in the event of a research-related injury to the subject.
(8) A statement that participation is voluntary, that refusal to participate will involve no penalty or loss of benefits to which the subject is otherwise entitled, and that the subject may discontinue participation at any time without penalty or loss of benefits to which the subject is otherwise entitled.
(b) Additional elements of informed consent. When appropriate, one or more of the following elements of information shall also be provided to each subject:
(1) A statement that the particular treatment or procedure may involve risks to the subject (or to the embryo or fetus, if the subject is or may become pregnant) which are currently unforeseeable.
(2) Anticipated circumstances under which the subject’s participation may be terminated by the investigator without regard to the subject’s consent.
(3) Any additional costs to the subject that may result from participation in the research.
(4) The consequences of a subject’s decision to withdraw from the research and procedures for orderly termination of participation by the subject.
(5) A statement that significant new findings developed during the course of the research which may relate to the subject’s willingness to continue participation will be provided to the subject.
(6) The approximate number of subjects involved in the study.
(c) When seeking informed consent for applicable clinical trials, as defined in 42 U.S.C. 282(j)(1)(A), the following statement shall be provided to each clinical trial subject in informed consent documents and processes. This will notify the clinical trial subject that clinical trial information has been or will be submitted for inclusion in the clinical trial registry databank under paragraph (j) of section 402 of the Public Health Service Act. The statement is: “A description of this clinical trial will be available on http://www.ClinicalTrials.gov, as required by U.S. Law. This Web site will not include information that can identify you. At most, the Web site will include a summary of the results. You can search this Web site at any time.”
(d) The informed consent requirements in these regulations are not intended to preempt any applicable Federal, State, or local laws which require additional information to be disclosed for informed consent to be legally effective.
(e) Nothing in these regulations is intended to limit the authority of a physician to provide emergency medical care to the extent the physician is permitted to do so under applicable Federal, State, or local law.
xviii Fact Sheet, Pfizer EUA, https://www.fda.gov/media/144414/download
xix Bridges v. Houston Methodist Hospital. Case 4:21-cv-01774 Filed 6/12/21 in TXD. Available at https://www.govinfo.gov/content/pkg/USCOURTS-txsd-4_21-cv-01774/pdf/USCOURTS-txsd-4_21-cv-01774-0.pdf



